Izmaylov Research Group

Research
The main efforts of our group are directed toward developing electronic structure and quantum dynamics methods on classical and quantum computers to obtain detailed understanding of processes involving simultaneous changes in electronic and nuclear states. Such processes constitute crucial steps in many areas of fundamental and technological importance: solar energy conversion, UV-light DNA damage and repair, operation of MRI contrast agents, catalysis at surfaces, and general surface chemistry.
Overview
Ongoing projects
When two or more electronic surfaces approach each other the Born-Oppenheimer approximation breaks down and description of nuclear dynamics needs to account for possible switching between multiple electronic surfaces. In molecules that have more than 2 atoms, very often electronic surfaces cross forming energy degenerate regions. One of the most common intersection motifs is a so-called conical intersection. 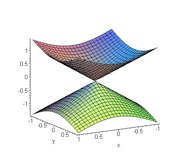 Conical intersections are very common participants of photochemical processes, one of the most well known is a vision process that starts from photo-isomerization of the retinal chromophore in the rhodopsin protein.
Conical intersections are very common participants of photochemical processes, one of the most well known is a vision process that starts from photo-isomerization of the retinal chromophore in the rhodopsin protein.
Interestingly, conical intersections not only create efficient channels between electronic surfaces but also impose special boundary conditions on electronic and nuclear wave-functions: a simple example is that if we go around the conical intersection both wave-functions must change their signs. In this series of projects we have two main themes: First, what are the consequences of this 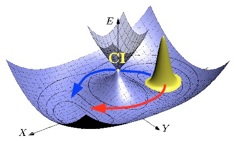 special boundary conditions for nuclear dynamics? Second, how to implement these boundary conditions efficiently in numerical simulations of chemical systems? Following the first theme we found that conical intersections can significantly slow down and even completely quench tunneling dynamics due to destructive interference. Further exploration of consequences of boundary conditions on nuclear dynamics as well as finding efficient techniques to account for them is an active area of research in our group.
special boundary conditions for nuclear dynamics? Second, how to implement these boundary conditions efficiently in numerical simulations of chemical systems? Following the first theme we found that conical intersections can significantly slow down and even completely quench tunneling dynamics due to destructive interference. Further exploration of consequences of boundary conditions on nuclear dynamics as well as finding efficient techniques to account for them is an active area of research in our group.
Representative publications:
-
1)I. G. Ryabinkin and A. F. Izmaylov, Geometric phase effects in dynamics near conical intersections: Symmetry breaking and spatial localization, Phys. Rev. Lett., 111, 220406 (2013)
-
2)I. G. Ryabinkin, L. Joubert-Doriol, and A. F. Izmaylov When do we need to account for the geometric phase in excited state dynamics? J. Chem. Phys. 140, 214116 (2014)
-
3)R. Gherib, I. G. Ryabinkin, and A. F. Izmaylov Why do mixed quantum-classical methods describe short-time dynamics through conical intersections so well? Analysis of geometric phase effects, J. Chem. Theory Comp., 11, 1375 (2015)
-
4)A. F. Izmaylov, J. Li, and L. Joubert-Doriol, Diabatic definition of geometric phase effects, J. Chem. Theory Comp. 12, 5278 (2016)
-
5)L. Joubert-Doriol, J. Sivasubramanium, I. G. Ryabinkin, and A. F. Izmaylov, Topologically correct quantum nonadiabatic formalism for on-the-fly dynamics, J. Phys. Chem. Lett. 8, 452 (2017)
YouTube videos:
2. Geometric phase effects in nonadiabatic dynamics
3. Method development for energy and charge transfer in organic molecules
Charge and energy transfers are at the heart of efficient solar energy harvesting and utilization. They also involve non-adiabatic dynamics involving several electronic states. In this project we develop new methods to model and understand these processes in middle size organic molecules  that can be seen as potential dye components of semiconductor cells or building blocks for organic photovoltaics. There are three main
that can be seen as potential dye components of semiconductor cells or building blocks for organic photovoltaics. There are three main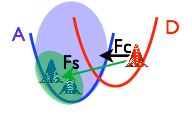 development branches: 1) perturbative non-equilibrium methods that start with diabatic model Hamiltonians built from first-principles electronic structure calculations [1], 2) on-the-fly wave-packet simulations with the perturbative spawning technique [2], 3) quantum-classical dynamics with the local energy operator approach [3].
development branches: 1) perturbative non-equilibrium methods that start with diabatic model Hamiltonians built from first-principles electronic structure calculations [1], 2) on-the-fly wave-packet simulations with the perturbative spawning technique [2], 3) quantum-classical dynamics with the local energy operator approach [3].
Representative publications:
-
1)A. F. Izmaylov, Perturbative Wave-packet Spawning Procedure for Non-adiabatic Dynamics in Diabatic Representation, J. Chem. Phys., 138, 104115 (2013)
-
2)L. Joubert-Doriol, I. G. Ryabinkin, and A. F. Izmaylov Non-stochastic matrix Schrödinger equation for open systems, J. Chem. Phys. 141, 234112 (2014)
-
3)J. Nagesh, A. F. Izmaylov, and P. Brumer An efficient implementation of the localized operator partitioning method for electronic energy transfer, J. Chem. Phys. 142, 084114 (2015)
-
4)J. Nagesh, M. J. Frisch, P. Brumer, and A. F. Izmaylov, Localized operator partitioning method for electronic excitation energies in the time-dependent density functional formalism, J. Chem. Phys. 145, 244111 (2016)
-
5)A. F. Izmaylov and L. Joubert-Doriol, Quantum Nonadiabatic Cloning of Entangled Coherent States, J. Phys. Chem. Lett. 8, 1793 (2017)
Metallic surfaces provide unprecedented capabilities for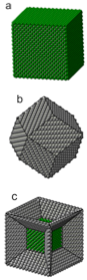 supporting, assembling, investigating, controlling molecular structures and chemical reactions. They are perfect for manipulation of matter on an atomic and molecular scale to build more efficient
supporting, assembling, investigating, controlling molecular structures and chemical reactions. They are perfect for manipulation of matter on an atomic and molecular scale to build more efficient 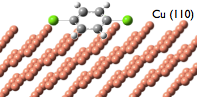 molecular-based devices (e.g., transistors, diodes, nanocircuits), nanoparticle heterogeneous catalysts, and metamaterials. In this project we develop new computational techniques and apply standard tools (mainly density functional theory) to do first principle modeling of molecules adsorbed on nanoparticles of different shapes (e.g., Pd) and Scanning Tunneling Microscope (STM) induced chemical reactions on surfaces of regular metals (e.g., Cu(110)). The main questions which are addressed: 1) What is a minimal adequate representation of electronic states of a molecule on a metallic surface? 2) What is the interplay between electronic and nuclear subsystems? 3) How does surface structure affect chemical reactions on metallic surfaces?
molecular-based devices (e.g., transistors, diodes, nanocircuits), nanoparticle heterogeneous catalysts, and metamaterials. In this project we develop new computational techniques and apply standard tools (mainly density functional theory) to do first principle modeling of molecules adsorbed on nanoparticles of different shapes (e.g., Pd) and Scanning Tunneling Microscope (STM) induced chemical reactions on surfaces of regular metals (e.g., Cu(110)). The main questions which are addressed: 1) What is a minimal adequate representation of electronic states of a molecule on a metallic surface? 2) What is the interplay between electronic and nuclear subsystems? 3) How does surface structure affect chemical reactions on metallic surfaces?
Representative publications:
-
1)A. Klinkova, P.V. Cherepanov, I. G. Ryabinkin, M. Ho, M. Ashokkumar, A. F. Izmaylov, D. V. Andreeva, E. Kumacheva, Shape-dependent Interactions of Palladium Nanocrystals with Hydrogen, Small, 12, 2450 (2016)
-
2)I. G. Ryabinkin and A. F. Izmaylov, Mixed quantum-classical dynamics using collective electronic variables: A better alternative to electronic friction theories, J. Phys. Chem. Lett. 8, 440 (2017)
-
3)I. Loaiza and A. F. Izmaylov, On the Breakdown of the Ehrenfest Method for Molecular Dynamics on Surfaces 149, 214101 (2018) arXiv:1809.03829
4. Modeling Chemical Reactions on Metallic Surfaces and Nanostructures
1. Quantum computing
The electronic structure problem is at the heart of our understanding of atomic and molecular electronic properties, which are so crucial for the rational molecular and material designs. Unfortunately, the computational cost of the electronic structure problem scales exponentially with the system size and presents a great challenge for a classical computer. On the other hand, there is a hope to overcome the exponential scaling by engaging a quantum computer. One of the main practical difficulties remains maintaining large enough number of qubits in a coherent superposition state entangling several particles. Another issue is related to reformulating the electronic structure problem for the quantum computer.
Recently, the Variational Quantum Eigensolver (VQE) method was developed to solve the electronic structure problem on a quantum computer. However, VQE is insufficient for accessing any molecular state but the ground state of an ionic form with the lowest electronic energy. For example, in the case of the H2 molecule, a potential energy surface of H2+ cannot be obtained (even though it is the simplest molecular problem with a single electron), because the cation is higher in energy than the neutral form. In our work, we fully resolve this embarrassing problem by proposing a methodology for constrained search [1]. The constrained search adds restrictions on the number of electrons and their spin in the state of interest.
Another common problem 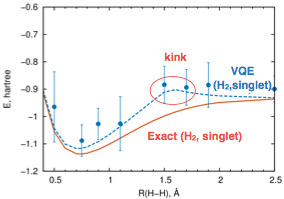 of VQE was that the dependence of the electronic energy from the nuclear configuration (i.e. the potential energy surface) exhibits kinks, the points with different slopes on different sides (left figure). These kinks, in many cases, are artificial and can be detrimental for investigating dynamical and equilibrium properties of molecules. The origin of this problem is the absence of necessary physical constraints in the conventional VQE scheme, which leads to symmetry breaking resulting in switching of the symmetry of the lowest electronic state. Imposing spin and number of electrons constraints ironed out all kinks in simulated potential surfaces (figure below).
of VQE was that the dependence of the electronic energy from the nuclear configuration (i.e. the potential energy surface) exhibits kinks, the points with different slopes on different sides (left figure). These kinks, in many cases, are artificial and can be detrimental for investigating dynamical and equilibrium properties of molecules. The origin of this problem is the absence of necessary physical constraints in the conventional VQE scheme, which leads to symmetry breaking resulting in switching of the symmetry of the lowest electronic state. Imposing spin and number of electrons constraints ironed out all kinks in simulated potential surfaces (figure below).
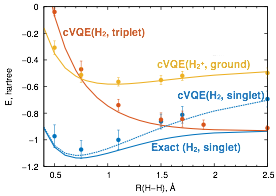
We demonstrate the capacity of our constrained VQE (cVQE) methodology by simulating ground states of the H2 molecule and its cation with 4 qubits on the 19Q-Acorn quantum processor. Our method features a low gate count with total number of gates linearly proportional to the number of qubits used. This enables us to treat even larger molecules, such as H2O (8 qubits and close to 200 non-commuting Pauli terms in the Hamiltonian)[1].
Representative publications:
-
1)I. G. Ryabinkin, S. N. Genin, A. F. Izmaylov, Constrained variational quantum eigensolver: Quantum computer search engine in the Fock space, J. Chem. Theory Comput. 15, 249 (2019) arXiv:1806.00461
-
2)I. G. Ryabinkin, S. N. Genin, A. F. Izmaylov, Relation between fermionic and qubit mean fields in the electronic structure problem, J. Chem. Phys. 149, 214105 (2018) arXiv:1806.00514
-
3)I. G. Ryabinkin, T. C. Yen, S. N. Genin, A. F. Izmaylov, Qubit coupled cluster method: A systematic approach to quantum chemistry on a quantum computer, J. Chem. Theory Comput. 14, 6317 (2018) arXiv:1809.03827
-
4)A. F. Izmaylov, T. C. Yen, and I. G. Ryabinkin, Revising measurement process in the variational quantum eigensolver: Is it possible to reduce the number of separately measured operators? Chem. Sci. 10, 3746 (2019) arXiv:1810.11602
In the news: