NMR
NMRs are an extremely versatile tool capable of determining structures of unknown compounds as well as their molecular interactions with other macromolecules. Traditionally, exposure to NMRs at an undergraduate level is rare due to its high costs and the complexity of operation. Over the years, user friendly operating software has been developed to reduce the difficulty of operating an NMR instrument.
Overview
All charged nuclei with a spin property create its own magnetic field; therefore are capable of interacting with an external magnetic field (B0). In the absences of (B0), these nuclei adopt random orientations. However, when an external magnetic field is applied, the nuclei align and the system splits into different spin states. For commonly observed nuclei such as 13C, 1H, 31P and 15N, they split into two energy levels: lower energy spin states are parallel with B0 while higher spin states are anti - parallel with B0. The samples are irradiated with a radio frequency pulse, exciting the nuclei from their low energy states to their high energy state. As the system relaxes, an NMR signal or spectrum is acquired. The experimental NMR spectrums can then be compared to the literatures for identifying different functional groups. (Note: Most NMR data are conveniently organised into tables for the purpose of identification)
Nuclear Spins
Electrons, protons and neutrons (called nucleons) are “active” spins. Nuclei with a non – zero spin value are said to be NMR active and therefore signals can be observed in an NMR experiment. The most common nuclei used in an NMR experiments are spins ½ (1H, 13C, 31P, and 19F), but some nuclei observed have a spin quantum number of 1 (2H).
Information seen on the NMR Spectrum
A bare nucleus will experience the full strength of an applied B0 field. However, in most cases,
the nucleus is surrounded by an electron cloud. Since electrons can generate their own magnetic field,
it will shield the nucleus from the full effect of the applied B0 field. The degree of this electron
shielding is a function of the property of the nuclei itself and its chemical environment. This electron
shielding effect is termed the chemical shifts and it is expressed as part per million (ppm).
The chemical shift scale is a reflection of the chemical environment around the nuclei. For
instance, the nucleus will be de-shielded if it is attached to a highly electron withdrawing group.
Therefore, it will experience more of the applied B0 field and more energy is required to excite the
nucleus, resulting in the signal being located in the higher ppm range or downfield. Since chemical
shifts are arbitrary, the values have to be standardized to an external reference. In this case,
the standard reference compound is tetramethylsilane (TMS) which has an assigned chemical shift of 0ppm.
The peak position in an NMR spectrum informs us of the origin of the signal: the presence of specific
structural groups. By comparing experimental results with literature values, which are conveniently
organised in Tables 1 and 2 1, chemical structures can be deduced. An extensive list of proton as well
as carbon NMR peaks can be found at, http://www.chem.wisc.edu/areas/organic/index-chem.htm. For
neighbouring nuclei with different chemical shifts values, they interact with each other through bond
(J-coupled) and gives raise to distinctive splitting patterns. These patterns provide information
of the neighbouring spins such that chemical structure can be deduced.
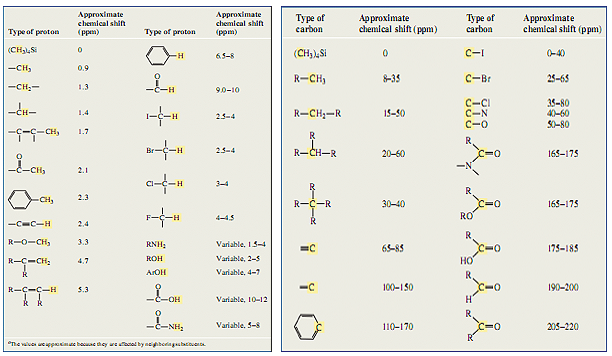
Table 1 Literature ppm of proton NMR Table 2 Literature ppm of carbon - 13 NMR
The number of chemically distinct neighbouring NMR active nuclei determines the splitting patterns observed in an NMR spectrum. Since these nuclei coupled with each other, they shared the same coupling constants. Importantly, coupling between nuclei with distance, therefore, the strongest coupling is observed between two nuclei directly bonded (one – bond coupling), followed by coupling between nuclei separated by two bonds (two – bond coupling). The magnitude of this coupling is expressed through coupling constant, J. The number of bonds is indicated in superscript in front of the J (Ex. 1J = one – bond coupling). The nuclei involved in coupling are represented with a subscript after J (Ex. 1J¬HP means one – bond coupling between H and P)
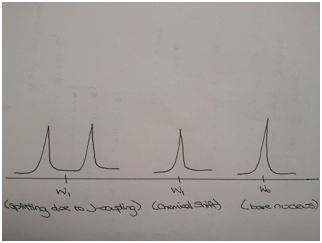
Figure 1 Effects of J - coupling and chemical shift on the bare nucleus
Magnets
The magnet is an essential part of the NMR instrument, since it is responsible for producing the main magnetic field (B0). In general, there are 3 types of magnets, permanent magnets, electromagnets, and superconducting solenoids.
Permanent magnets are used for low – field (0.7 – 2.1T) continuous wave (CW) instruments. CW instruments are older and measures the response of sample as radio frequency (RF) is changed. These magnets are temperature sensitive and much heavier. Electromagnets are no longer used in modern instruments. These magnets use electric current to generate the magnetic field. Superconducting solenoids are most commonly used in modern FT – based instruments and provide field strengths as large as 23T. As a superconductor, a wire (Nb – Sn or Nb – Ti) are wound up into a multi – turn coil. These magnets are favoured since its superconductivity provides a state with negligible resistivity. These magnets must be kept cool in order to maintain superconductivity but increase in temperature as B0 increases. Therefore, these magnets are submerged in liquid helium which is surrounding by liquid nitrogen in order to keep the helium cool. Both the helium and the nitrogen must be refilled, but helium only requires refilling every 3 months – 1 year depending on the instrumental design. Nitrogen on the other hand must be refilled much more frequently.
Locking and Shimming
Ideally, the magnets used in NMR should produce a constant and homogenous field, but in reality, this magnetic field drifts over time. Therefore, corrections must be made in order to optimize results obtained. This drift can be accounted for by locking and shimming the instrument. In order to lock the instrument, a nucleus, usually deuterium, is used as a reference. Various deuterated solvents, CDCl3, DMSO-d6, C6D6, can be used for locking purposes The nucleus is constantly monitored at its frequency for a given field strength, if changes in the signal are seen, they are reported to the instrument which can then correct for the drift. Prior to any measurements, the instrument must be locked. If a new sample with a different solvent is introduced in the instrument, relocking is necessary, but if the solvent in the new sample is the same as the previous sample, locking can be skipped.
Shim coils are pairs of wire loops controlling low currents pass. The shim coils produce a magnetic field which compensates for any inhomogenities in the permanent field. After the instrument is locked, the shim magnetic fields must be adjusted, this process is called shimming. Whenever a new sample is introduced in the instrument, shimming must be repeated. The shimming process is complete when the lock level is maximized. If the instrument was not shimmed properly, the peaks observed on the spectrum should resemble those seen in Figure 2.
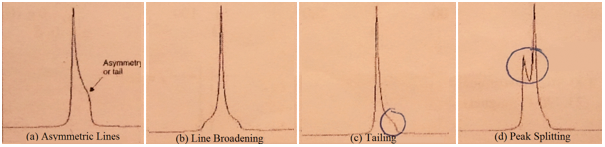
Figure 2 Abnormalities seen in peaks due to inadequate shimming.
Probe The NMR probe is capable of emitting and receiving RF signals. It contains the RF coil, sample spinner, temperature controlling circuitry, and gradient coils. The sample is placed within the RF coil of the probe which produces the necessary RF frequency for exciting the spins and detection of NMR signals from the sample. The probe is also capable of controlling the temperature of sample above and below the room temperature which allows for analysis of the kinetic processes. Probes are tuned to emit frequencies characteristic for different nuclei in a given magnetic field. Modern probes can cover several NMR active nuclei and allows for more advanced multi-dimensional experiments to be run. Types of NMR Analysis
A single resonance NMR spectrum (1D NMR) and double resonance spectrums (2D NMR) can be used for structure determination. A 1D NMR, such as 1H or 13C NMR, applies a single RF pulse (B1) to the sample of interest, resulting in spectra such as those seen in Figure 3 for 1H NMR.1 Also, for 1D 13C, 1H decoupling is used such that a singlet is observed with greater sensitivity. NMR For a double resonance heteronuclear correlation experiment (1H-X (X = 13C or 15N)), pulse sequences such as HSQC (Heteronuclear Single Quantum Coherence), HMQC (Heteronuclear Mulitple Quantum Coherence), or HMBC (Heteronuclear Mulitple Bonds Coherence) are used. (Note: For more information on 2D NMR please consult this website, http://www-keeler.ch.cam.ac.uk/lectures/2d_letter.pdf)
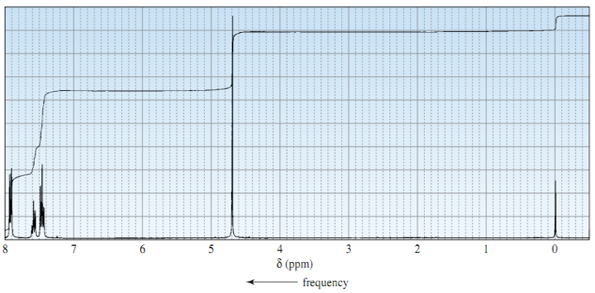
Figure 3 Example of a 1H NMR spectrum
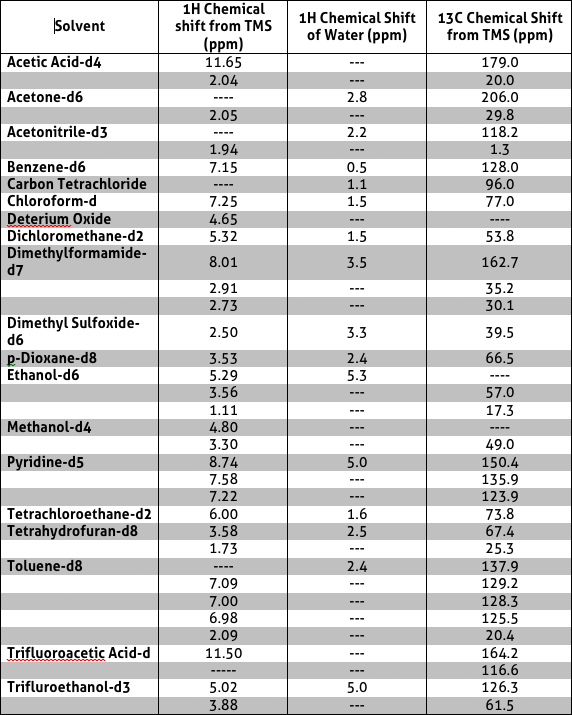
Sample Preparation In order to utilise the NMR to obtain desired information regarding the sample of interest, the sample must first be prepared utilising appropriate NMR solvents in a NMR tube. A list of appropriate solvents to use for NMR sample preparation is listed in Table 3.2 deutrated solvents are most commonly used since they are inert and have minimum 1H background where residual solvent peaks are observed. Table 3 includes the chemical shifts of each solvent. The most commonly used solvents available in the NMR centre are Deuterium Oxide, CDCl3, DMSO-d6 and C6D6.
The NMR tubes available in the NMR centre are New Era NMR tubes. These tubes are made of glass and will
break/crack when dropped. Therefore, these tubes must be handled with extra care. If the NMR tubes are
chipped or cracked, DO NOT utilise these tubes and dispose them in the broken glass bins in the lab.
When analysing solid samples, only ~ 10mg of sample should be used along with 500 to 800 µL of
deutrated solvent. When preparing liquid samples, approximately 50-100 µL of sample will be used
along with 500 to 800 µL of deutrated solvent.
(Note: Do not over dilute your samples (i.e. 100 times)
since NMR is an insensitive technique)
Overall, the samples prepared should not occupy more than ½ of
the NMR tube. Once the sample is prepared, cap and label the NMR tubes. A depth gauge, provided in
the NMR center, is used to measure the appropriate height of the sample. The NMR utilised in the
lab is a Bruker Avance 3, 500 MHz NMR with a 5mm BBFO probe. Since the NMR is a fairly strong
magnet, electronics should not be brought near this instrument, if required, electronics, credit
cards etc. can be left in the sample preparation room. Remember the slot number your sample is
in for quick retrieval of sample and data later on!
Table 3 Solvents appropriate for NMR sample preparations as well as possible residual peak visible on the spectrum
References
1 Bruice, P.Y. Organic Chemistry, 5th ed.; Pearson Prentice Hall: New Jersey, 2007
2 Spectral Data Services. NMR Solvent Chemical Shifts Table. http://www.sdsnmr.com/cs_table.html. (accessed June 5, 2011)